How many types of FDA IND application?
There are multiple types of IND application. Generally, your IND will be either 1) commercial or 2) research. It depends on whether it will be used to support marketing application afterwards.
There are then three IND ‘types’ underneath this broad categorization:
- Investigator IND, for investigation of an unapproved drug. This is the most common IND type.
- Emergency use IND, for quick authorization of a drug for use in an emergency situation (such as the COVID-19 pandemic) when no alternative treatment exists
- Treatment IND, for drugs already in the clinical stage which are already proved effective in treating serious conditions
When should you start your IND process?
The IND stage begins once the pre-clinical activities are concluded. That means you should:
- Be confident in the strength and integrity of your product performance, and ready to take it into the human testing stage.
- Have sufficient proof-of-concept data to present to the FDA to attest to your product’s safety and efficacy so far.
What data and information need submitted for IND application?
- Toxicology and pharmacology data from animal testing
- Storage and stability data
- Manufacturing information showing how you intend to manufacture your product at scale and with sufficient quality
- Clinical research plans and clinical protocols to assess potential risk to humans
- Target clinical indication to contextualize your submission
IND submissions
What happens after the IND submission?
Your collected data should be submitted in triplicate to the FDA alongside three supplementary forms.The FDA then has 30 days to review and approve your product. In the case of approval, or simply a lack of rejection in the 30-day timeframe, your product has the green light to progress to the clinical testing stage and may be transported to clinical investigators in different states.
What are the most common reasons for IND data pack rejections:
- Submission mistakes: missing documents or incorrect eCTD formats
- Disorganized and/or unclear data that the FDA simply cannot process and digest within 30 days
- Unfocused and irrelevant data that doesn’t support your clinical protocol or adequately explain potential risks
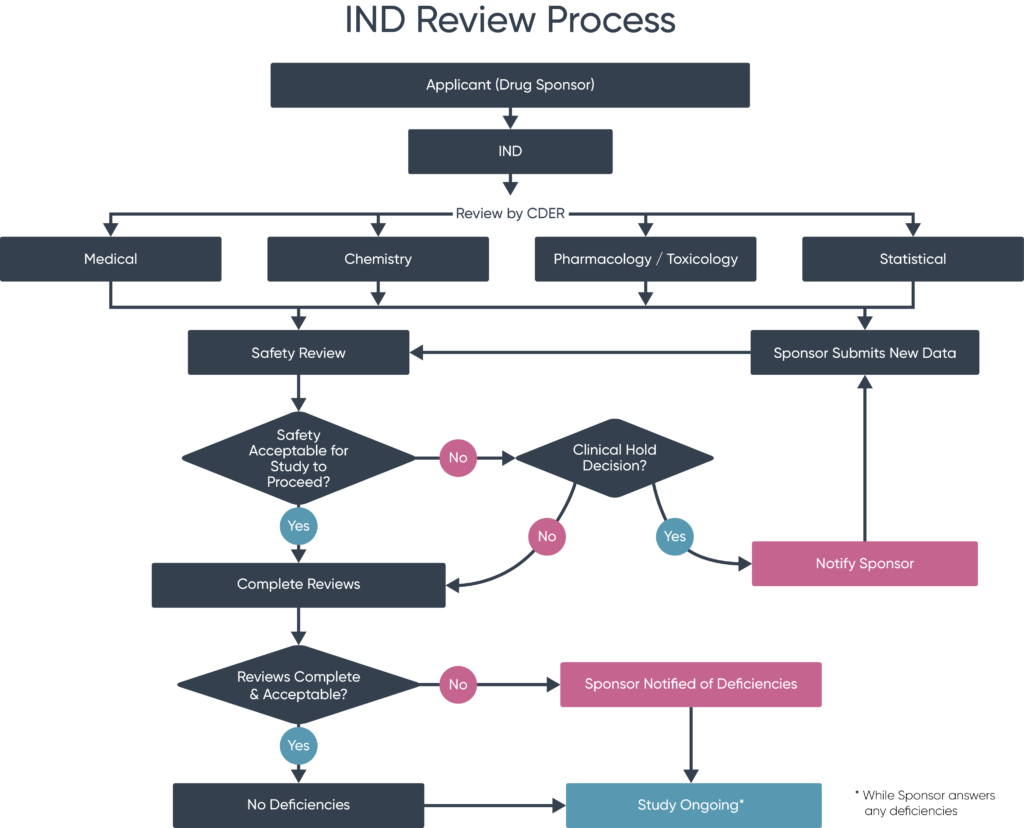
How does the IND Review process work?